
Responsible Science is the Future of our Planet
At Gaia Science, we believe that advancing technology responsibly is key to protecting our planet and improving lives through sustainable scientific i
Request a Quote
Empowering Science with Innovation
Delivering advanced laboratory solutions and cutting-edge technologies to universities, research institutions, and industries worldwide.
Request a Quote
Global Leader in Automated Biogas & Biodegradability Testing
Automatic Methane Potential Test System | Novel Gas Volume and Flow Meter | Biodegradability Assessment System | Respiration & Fermentation Tester
Learn MoreOur Products
We provide excellent laboratory equipment to our customers


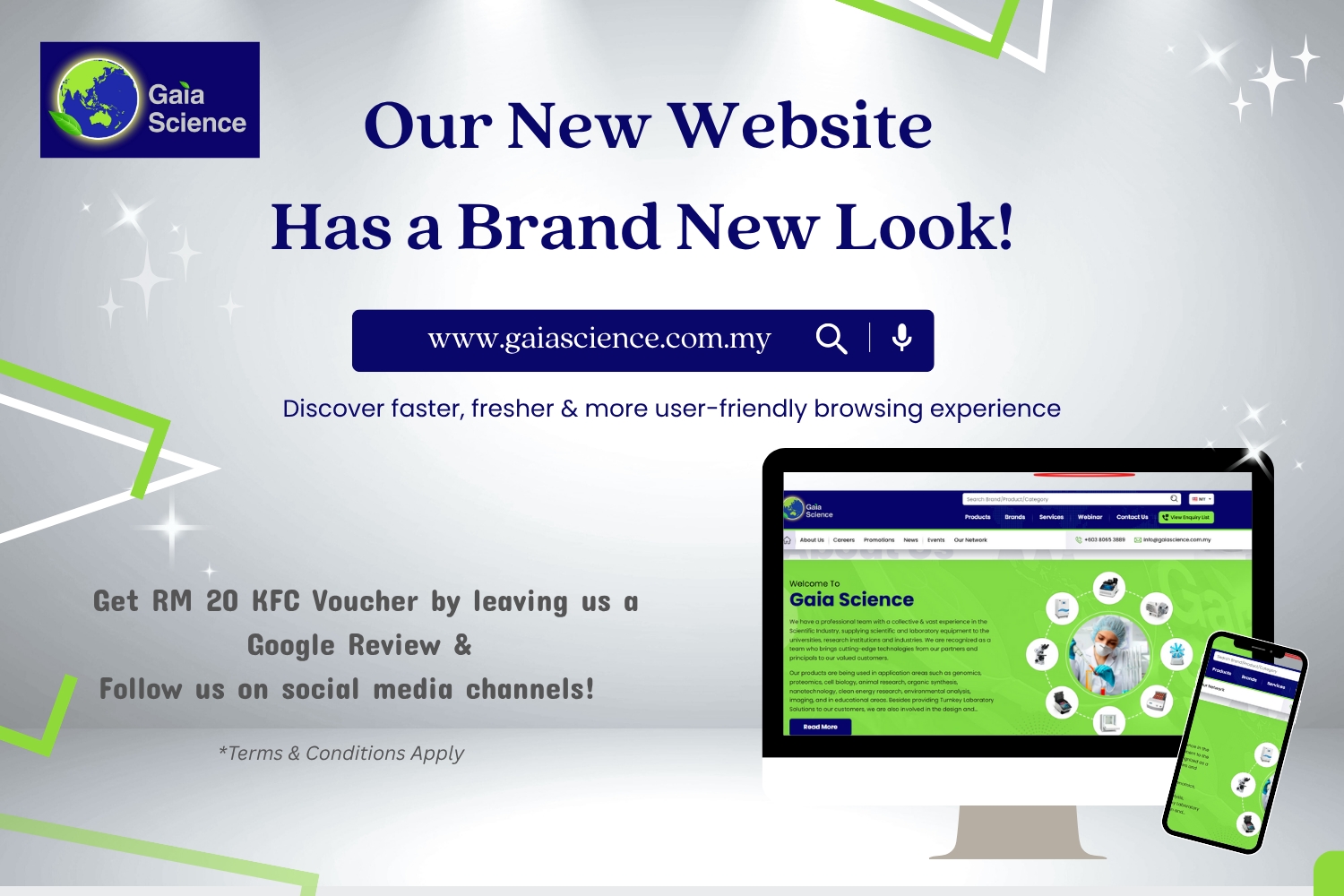
Welcome To
Gaia Science
We have a professional team with a collective & vast experience in the Scientific Industry, supplying scientific and laboratory equipment to the universities, research institutions and industries. We are recognized as a team who brings cutting-edge technologies from our partners and principals to our valued customers.
Our products are being used in application areas such as genomics, proteomics, cell biology, animal research, organic synthesis, nanotechnology, clean energy research, environmental analysis, imaging, and in educational areas. Besides providing Turnkey Laboratory Solutions to our customers, we are also involved in the design and manufacturing of bioengineering products such as mobile laboratories, modular BSL-3 laboratories, tissue trimming tables, necropsy tables and etc with international partners.
Bridging state-of-the-art laboratory products to the scientific community is our aim and providing service excellence is our goal. We see the scientific community as our partner in promoting scientific and technological advancement for a better world and better quality of life for all.

For any queries or support, kindly get in touch with us.
Ongoing Promotions
Enjoy exclusive deals and special savings happening right now for a limited time.
Latest News
Explore recent announcements, upcoming events, and highlights from the world of science.

World Renowned Brands












Featured Products














We're Here to Help
Fill out the form below and our team will get back to you shortly with the support you need.